You probably already know the feeling. A formulation misses its target by a narrow margin, so the team adjusts one additive, reruns the experiment, waits for characterization, then discovers the change fixed one property and broke two others. Weeks disappear into a loop of educated guesses, fragmented lab notes, and half-comparable test results.
That's still how a lot of materials R&D operates. It works, but it's slow, expensive, and hard to scale across formulation space, process windows, and competing performance requirements. The problem isn't that scientists lack intuition. The problem is that modern materials problems have too many interacting variables for intuition alone to search efficiently.
Machine learning materials science changes that when it's implemented well. Not as a magic button, and not as a replacement for bench work. It changes the operating model by helping teams learn from historical experiments, rank the next most informative trials, and connect structure, process, and property faster than manual analysis can.
This shift isn't hypothetical anymore. A 2024 review in Chemical Materials reported that the number of studies applying machine learning to materials science has been growing at approximately 1.67× per year over the past decade, showing that the field has moved from a niche research topic into a rapidly expanding mainstream approach for discovery and optimization (Chemical Materials review on machine learning growth in materials science). That matters because rapid publication growth usually signals something deeper than hype. It means methods are diversifying, datasets are improving, and teams are finding enough practical value to keep investing.
The companies that benefit most won't be the ones with the fanciest model names in a slide deck. They'll be the ones that solve the unglamorous implementation work: data plumbing, validation discipline, and organizational trust.
Table of Contents
Introduction The New R&D Paradigm
A formulation team spends six weeks adjusting resin ratios, cure conditions, and filler loading. The lab generates plenty of results, but half the context lives in spreadsheets, some in instrument files, and some in researchers' notebooks. The final review meeting produces a familiar conclusion. There is signal in the work, but nobody can say with confidence which variables drove the outcome or which experiment should come next.
That is the operating problem machine learning can improve in materials R&D. Its value starts with search efficiency. A good ML workflow helps teams cut down the candidate space, surface inconsistent results early, and rank the next experiments with more discipline than trial and error alone.
Why the old model strains under modern complexity
Industrial materials programs rarely target one property in isolation. Teams usually have to balance strength, viscosity, cure profile, thermal behavior, conductivity, stability, processability, cost, and manufacturability at the same time. Add supplier variability, scale-up effects, and differences in test methods, and the search space grows faster than most experimental programs can handle.
Many ML articles spend too much time on model types and too little on implementation. In practice, R&D groups usually stall before model selection becomes the main issue. The hard part is building a usable chain from composition and processing history to test conditions and measured outcomes, then keeping that chain consistent as projects evolve.
Practical rule: If your team cannot reliably answer “what exactly was tested, under which conditions, and how was the property measured?”, you do not have a modeling problem yet. You have a data definition problem.
This is also why adoption often disappoints on the first attempt. Teams buy software or run a pilot model, then discover that naming conventions differ by site, key process variables were never logged, and historical results cannot be compared because the test protocol changed midstream. The organizations that get value from materials ML treat data infrastructure, experiment design, and model operations as part of the same R&D system.
Why this shift is happening now
The field has moved past isolated proofs of concept. As noted earlier, publication activity has been rising quickly, and that growth has brought more benchmark datasets, better tooling, and more examples tied to real discovery and optimization work. For industrial teams, the bigger change is operational. Electronic lab records are more common, lab instruments export more structured data, and compute is cheap enough to support routine model retraining instead of one-off analysis.
Skepticism is still healthy. The better question is where ML creates repeatable advantage inside an R&D workflow, and what conditions make that advantage real.
Those conditions are demanding. Teams need data that is fit for the decision at hand, models matched to the size and shape of the dataset, and scientists who treat predictions as one input into an experimental program rather than a substitute for judgment. When those pieces are in place, the workflow changes in a practical way. Researchers stop screening broad sets of plausible options and start running tighter design, build, test, learn cycles with clearer rationale for each experiment.
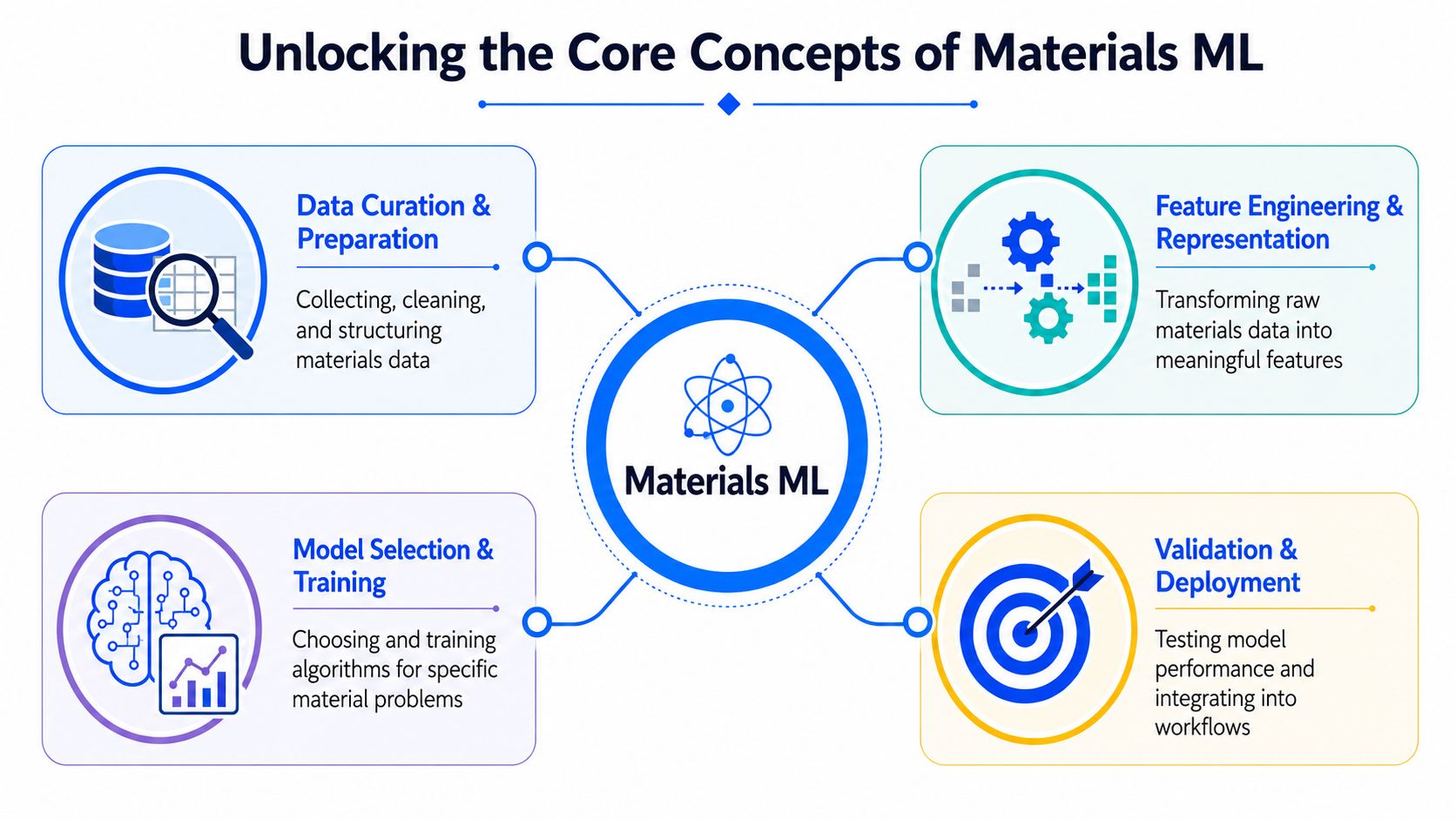
Unlocking the Core Concepts of Materials ML
Materials ML starts to make sense once you view it as an R&D decision system, not just a modeling exercise. In practice, the question is rarely “which algorithm should we use?” It is usually “what can we predict early enough, and reliably enough, to change the next experiment?”
That distinction matters in industrial settings. Teams are working with formulation tables, process logs, microscopy images, spectroscopy outputs, and property measurements collected for different purposes and under different conditions. The model has to turn that uneven record into predictions a scientist can act on.
How a model sees a material
A materials scientist sees polymer architecture, filler dispersion, phase behavior, grain boundaries, or defect chemistry. A model sees representations. Those representations might be hand-built descriptors from composition, structure-based fingerprints, embeddings learned from images or spectra, or feature sets that combine formulation variables with process conditions and test metadata.
That translation step is where many projects succeed or fail.
A useful workflow usually includes five parts:
- Data curation. Select experiments that are comparable, define labels carefully, and remove records that would confuse the learning task.
- Feature engineering. Convert raw materials information into inputs the model can use, whether numerical descriptors, categorical variables, graphs, images, or sequence-like data.
- Model training. Fit a model to map inputs to outcomes such as viscosity, conductivity, modulus, yield, lifetime, or pass/fail status.
- Validation. Test whether performance holds on unseen data that reflects real lab decisions, not just random splits that look good on paper.
- Deployment. Put the model inside an experimental workflow so it can rank candidates, flag risky trials, or suggest the next measurement.
Teams often spend too much time comparing architectures and too little time defining the prediction target, the decision threshold, and the acceptable error. In R&D, those choices determine whether a model saves a month of work or creates false confidence.
The tasks that matter in practice
In industry, materials ML usually supports a small set of recurring tasks:
- Property prediction estimates outcomes before synthesis or testing. This is often the fastest route to measurable value.
- Classification separates likely passes from likely failures, or stable systems from unstable ones, so teams can reduce low-probability experiments.
- Optimization searches across formulation and process variables to improve multiple properties at once.
- Inverse design starts with a target performance profile and proposes candidate materials, compositions, or processing windows.
- Interpretation helps scientists check whether the model is learning physically plausible relationships or exploiting artifacts in the dataset.
The trade-offs are real. A highly flexible model may improve headline accuracy but make it harder to explain why a candidate was selected. A simpler model may be easier to trust and maintain, especially when the dataset is small and the cost of a wrong recommendation is high. In many programs, the better choice is the model that fits the decision context, retrains cleanly, and gives the team a defensible basis for the next experiment.
As noted earlier, recent reviews have shown strong results across atomic, microstructural, and bulk property prediction tasks. That breadth is important, but it does not remove the implementation burden inside an R&D organization. A model only matters if scientists can check its reasoning, understand its uncertainty, and connect its output to an experimental plan.
A model that predicts well but cannot be checked against scientific intuition rarely earns trust in an R&D organization.
That is why explainability matters in materials ML. Scientists do not need a lecture on every algorithmic detail. They do need enough visibility to challenge the result, spot spurious correlations, and decide whether the recommendation deserves lab time. In successful teams, ML supports judgment. It does not replace it.
Fueling the Engine Data for Materials Science ML
Most failed ML projects in materials science don't fail because the algorithm was too simple. They fail because the data was inconsistent, incomplete, or impossible to align across sources. In this field, data is rarely abundant and tidy. It's usually small, sparse, expensive, and context-heavy.
A formulation result without mixing conditions may be useless. A microscopy image without sample history may mislead the model. A thermal measurement without instrument settings or operator notes may not be comparable to prior runs. Materials data carries process and provenance with it, and models inherit whatever ambiguity you leave unresolved.

Why materials data is different
In consumer software, you can often collect more user events and smooth out noise with scale. In materials R&D, every datapoint can represent a costly synthesis, a long test cycle, or a destructive characterization run. That changes how you design the project.
A major review notes that materials ML is especially effective in small-data regimes, and that model choice should prioritize sample efficiency, interpretability, reliability, and alignment with physical intuition. The same review notes that common evaluation practice relies on K-fold cross-validation or leave-one-out cross-validation, rather than a single train/test split (Nature review on machine learning in small-data materials settings).
That's an important operational point. If your dataset is limited, a casual split can give you a flattering result that vanishes in production. Better validation discipline won't make the model smarter, but it will make your decisions more honest.
What an AI-ready data backbone actually includes
An AI-ready materials dataset is not just a cleaned spreadsheet. It usually needs several layers of structure:
- Composition and identity data for monomers, additives, fillers, solvents, precursors, or processing aids.
- Process metadata such as order of addition, mixing energy, temperature profile, curing conditions, residence time, or pressure history.
- Measurement context including instrument type, test method, calibration status, sample geometry, and environmental conditions.
- Outcome variables that are consistently named, unit-normalized, and tied to actual sample lineage.
- Failure records rather than only “good” experiments. Negative results often carry the strongest learning signal.
Teams that do this well also create data governance rules early. They define naming conventions, unit standards, ontology choices, and ownership of curation. It's not glamorous work, but it determines whether later model outputs are actionable.
The fastest way to slow down an ML program is to let every lab group keep its own definition of the same property.
If you're leading implementation, resist the urge to wait for perfect data. Start with one problem where data quality is good enough, but invest immediately in structure that can scale. In real organizations, value usually appears when historical experiments become searchable, comparable, and reusable across teams. The model is only one beneficiary of that cleanup. Scientists benefit first.
Choosing Your Toolkit Common ML Models and Approaches
A team has six months of formulation data, three properties worth predicting, and one request from management: tell us what to test next. That is not the moment for the most advanced model on arXiv. It is the moment to choose a toolkit that fits the data you have, the decisions you need to make, and the level of trust your scientists will demand before they change an experimental plan.
Model choice in materials R&D is an implementation decision as much as a statistical one. The best-performing model in a benchmark can still fail in practice if it needs representations your lab cannot generate reliably, takes too long to retrain, or produces outputs your formulation chemists will not act on. Good teams start with the decision point. Screening, ranking, classification, surrogate optimization, inverse design, and uncertainty estimation each push you toward different model families.
What works when the dataset is limited
For many industrial programs, the first useful models are still conventional supervised methods:
| Model | Primary Use Case | Data Requirement | Interpretability | Best For |
|---|---|---|---|---|
| Random Forest | Property prediction, classification | Low to moderate | Moderate | Fast baselines on tabular experimental data |
| Gradient Boosting | Property prediction, ranking | Low to moderate | Moderate | Strong performance on structured datasets with nonlinear effects |
| Linear Models with engineered features | Screening, trend estimation | Low | High | Sanity checks and physically interpretable relationships |
| Gaussian Process models | Optimization, uncertainty-aware prediction | Low | Moderate | Sequential experimentation and active learning |
| Support Vector Machines | Classification, regression | Low to moderate | Lower | Well-bounded datasets with carefully engineered descriptors |
| Neural Networks | Complex nonlinear prediction | Moderate to high | Lower | Richer datasets where feature learning matters |
| Graph or composition-aware deep models | Materials property prediction | Moderate to high | Lower to moderate | Cases with useful structural or compositional representations |
| Generative models | Inverse design, candidate generation | High and problem-specific | Low | Proposing new candidates that still need filtering and validation |
Random forests and boosting models remain common for a reason. They train quickly, tolerate messy nonlinear relationships, and usually give a strong first answer on tabular lab data. In many industrial settings, that is enough to create value early.
Linear models still matter too. If a simple model with well-chosen descriptors performs almost as well as a more complex one, the simpler model often wins because scientists can inspect the drivers, challenge bad correlations, and connect the result back to known chemistry or process physics.
Gaussian processes deserve special attention in R&D settings where experiments are expensive. They are often less attractive for very large datasets, but they can be extremely useful when the primary goal is experiment selection rather than pure prediction accuracy. If the next batch of experiments costs weeks of lab time, calibrated uncertainty can matter more than squeezing out a small gain in cross-validation score.
Where newer deep learning models fit
Deep learning starts to earn its place when representation is the hard part. Crystal structures, molecular graphs, microstructure images, spectra, and sequence-like process records often contain patterns that hand-built descriptors miss or flatten too aggressively. In those cases, graph networks, attention-based models, or multimodal architectures can outperform classic approaches.
The trade-off is operational, not just technical. Deep models usually need more care in featurization, hyperparameter tuning, compute setup, and monitoring for drift. They also make failure analysis harder. When a random forest performs poorly, teams can often trace the issue to a missing variable, a bad split, or a thin region of the design space. When a deep model fails, diagnosis is slower and retraining is more expensive.
That is why strong baselines matter. In a production-minded workflow, advanced models should beat simpler alternatives under fair validation and justify their extra maintenance cost. If they do not, keep the simpler system.
A second lesson from recent materials ML work is that no single model is likely to cover every problem your organization cares about. The more realistic pattern is a modular stack: one model for property prediction, another for uncertainty, a rules layer for chemistry or processing constraints, and a ranking step that decides what reaches the lab. A recent review makes this point clearly while also stressing that these systems still need verification because black-box and statistical errors do not disappear at deployment (review on modular scientific materials models).
That modular view usually matches how successful R&D groups operate. A battery team might use gradient boosting for cycle-life screening, a graph model for electrode candidate embeddings, and Bayesian optimization to choose the next experiments. A polymers group might keep a linear benchmark for interpretability, use a tree ensemble for day-to-day ranking, and reserve a generative model for ideation after practical formulation constraints are encoded. The gain comes from model orchestration tied to lab decisions, not from chasing one universal architecture.
Choose the toolkit your team can run, audit, and improve. Then add complexity only when it changes experimental outcomes.
From Prediction to Discovery Real-World Applications and Workflows
The most useful way to think about ML in materials R&D is not as a standalone prediction engine, but as part of a design build test learn system. A model proposes. The lab verifies. The new data feeds back into the next round. Over time, the cycle becomes sharper and less wasteful.
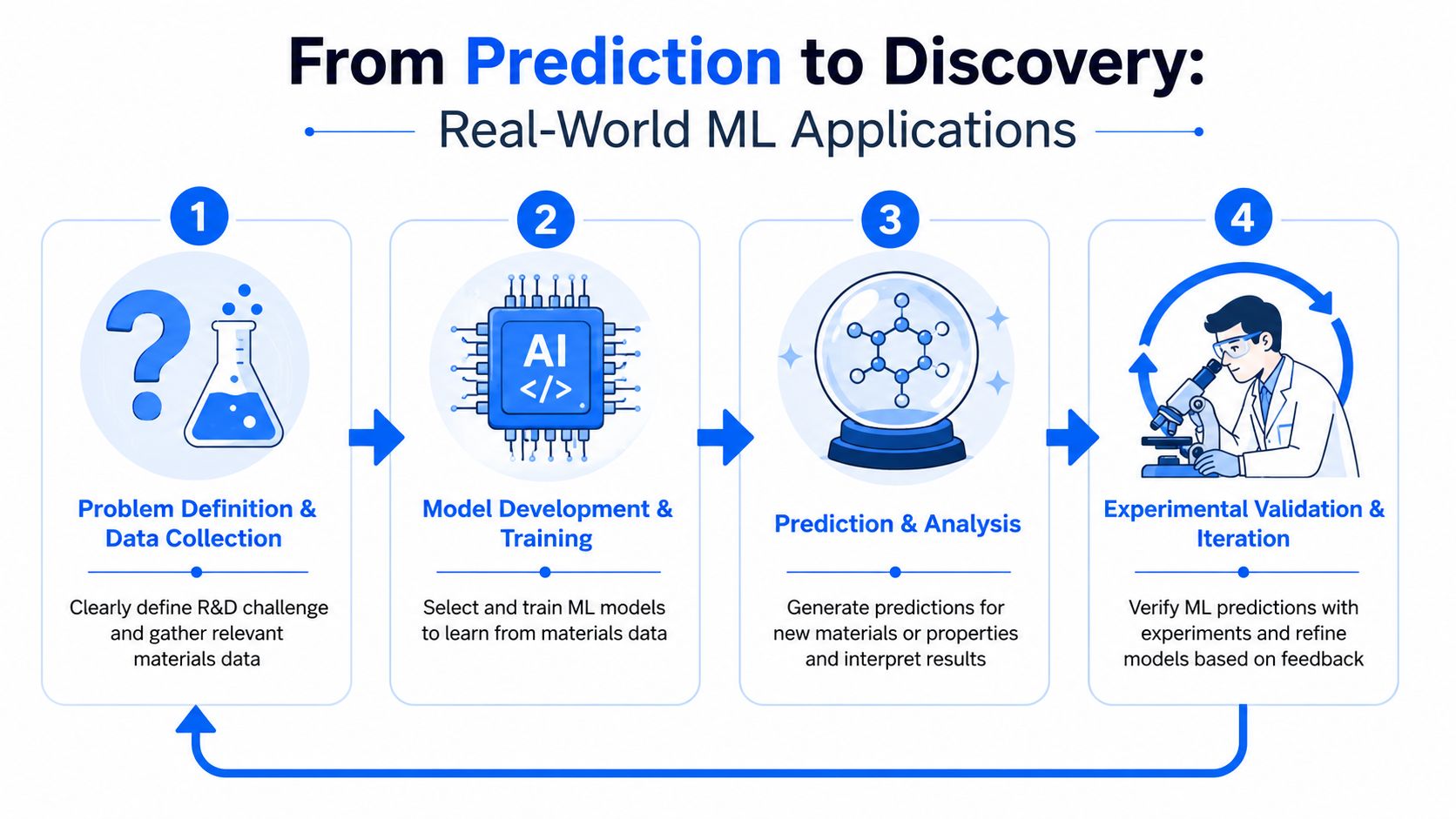
The modern design build test learn loop
In formulation work, that loop often starts with a familiar pain point. A team has historical recipes and test outcomes, but no reliable way to infer which variables matter most or which unexplored region is promising. ML can rank candidate formulations, highlight likely trade-offs, and reduce the number of experiments wasted on low-probability options.
In computational materials work, the impact can be even more dramatic. A review of ML in materials science reports that properly trained models can match DFT-level accuracy for property prediction, and that ML-augmented molecular dynamics methods such as DeePMD have enabled ab initio-quality simulations for systems exceeding 100 million atoms and timescales beyond 1 nanosecond (review on ML matching DFT accuracy and large-scale simulation). For R&D leaders, the practical meaning is simple: surrogate models can move some decisions earlier, before the team pays the full cost of high-fidelity computation or extensive lab work.
A short visual helps frame that workflow in operational terms.
Where teams see value first
The earliest wins usually come from problems with clear targets and repeatable measurements.
- Formulation optimization: Teams use historical composition and process data to predict likely property windows, then prioritize the next experimental batch.
- Virtual screening: Candidate materials can be filtered before synthesis or purchase, which reduces load on expensive downstream testing.
- Process-structure-property modeling: ML links operating conditions to morphology and final performance, which helps when scale-up changes outcomes in non-obvious ways.
- Anomaly detection: Models flag experiments that look inconsistent with prior knowledge, which can reveal bad inputs, failed runs, or measurement drift.
When ML works in the lab, it rarely removes experimentation. It changes which experiments are worth running next.
That distinction matters. Scientists still handle synthesis strategy, mechanistic reasoning, and final judgment. The model compresses the search space. It does not remove the need for domain expertise.
The strongest workflows also close the loop fast. Predictions should be stored with the actual experiment that tested them. Scientists should be able to see where the model was right, where it drifted, and which types of samples remain underrepresented. That feedback discipline turns machine learning from a slideware initiative into a working part of discovery.
Implementation in R&D Best Practices and Pitfalls
Most organizations don't struggle with the idea of AI anymore. They struggle with putting it into a workflow that bench scientists trust and managers can justify. At this juncture, machine learning materials science projects either become operational or stall in pilot mode.
A U.S. Department of Energy review identifies four barriers to broader adoption in materials science: cognitive resistance, lack of know-how, weak data infrastructure, and algorithm limits. The same review recommends standardization, uncertainty quantification, standardized instrument APIs, standardized high-throughput methods, and round-robin benchmarking to build trust (Department of Energy review on adoption barriers in materials ML).
The four barriers that usually stall adoption
Cognitive resistance shows up when chemists or materials engineers hear “black box” and assume the system will override judgment. That reaction is rational. Many ML projects have earned it by presenting predictions without provenance, uncertainty, or scientific context.
Lack of know-how appears on both sides. Data scientists may not understand assay variability, formulation constraints, or processing realities. Domain experts may not know how to frame a modeling problem, interpret validation results, or spot leakage.
Weak data infrastructure is the barrier I see most often. The company may have years of experiments, but they're trapped across spreadsheets, instrument files, slide decks, local drives, and partially filled ELNs.
Algorithm limits become obvious when leaders expect one model to solve every use case. Some problems are underdetermined. Some datasets are biased. Some targets are too noisy to support a dependable predictor.
What successful teams do differently
The best teams don't launch with a platform-first mindset. They launch with a decision-first mindset. They pick one expensive, recurring question where better ranking or screening would change behavior. Then they build around that.
A practical playbook looks like this:
- Start with a narrow, high-value pilot. Choose a problem with enough historical data, a measurable lab outcome, and a decision bottleneck the team already feels.
- Use interpretable baselines first. A simpler model with sensible drivers will often gain more adoption than a deeper model that performs slightly better but can't be explained.
- Treat uncertainty as a product feature. Scientists need to know when the model is extrapolating, not just what it predicts.
- Standardize before you scale. Property names, units, sample lineage, and test methods need consistent definitions.
- Benchmark against current practice. Compare the ML-assisted workflow to how the team already decides what to test next.
- Keep domain experts in the loop. They should challenge outputs, reject implausible suggestions, and help encode constraints.
One underused move is to avoid building everything from scratch. Depending on your stack, teams may combine internal data platforms, workflow tools, and domain-specific systems. For example, some organizations use platforms such as Polymerize to unify fragmented experimental data and apply explainable, domain-specific materials models within an R&D workflow. That can be sensible when the bottleneck is integration and execution rather than raw model experimentation.
If you're building a new initiative around this capability, external capital and ecosystem support may also matter. Corporate venture teams and startup operators sometimes look at the broader funding environment before launching tooling or partnership efforts, and resources like this guide to search for US machine learning investors can help map the market around AI infrastructure and applied ML.
Don't wait for perfect data, but don't pretend messy data is harmless. Clean enough to decide is the threshold. After that, improve iteratively.
Common pitfalls are predictable. Teams hide failed experiments, which weakens the training signal. They judge models on random splits that don't reflect future use. They deploy a predictor without retraining plans. Or they treat the first good result as proof the system is production-ready. In materials R&D, trust grows from repeated, transparent usefulness.
Conclusion The Future of Materials Discovery is Collaborative Intelligence
The strongest argument for machine learning in materials science isn't that it makes R&D automatic. It's that it makes R&D more deliberate. Instead of relying on memory, scattered notes, and slow iteration alone, teams can combine historical evidence, predictive models, and experimental judgment in a tighter loop.
That only works when the fundamentals are respected. Data has to be curated with scientific context. Models have to be selected for the problem, not for novelty alone. Validation has to reflect real operating conditions. And implementation has to address human trust as seriously as technical performance.
The future here is not human versus machine. It's collaborative intelligence. Models handle pattern recognition across a search space too large for any team to inspect manually. Scientists bring mechanism, skepticism, creativity, and the ability to decide what should be tested, not just what could be predicted.
That partnership is where the true advantage lies. The organizations that build it well will learn faster from every experiment, waste less effort on low-value trials, and move from broad trial-and-error toward targeted discovery. In polymers, chemicals, and advanced materials, that shift can reshape how programs are planned, how scale-up risk is managed, and how quickly new products reach reality.
Machine learning materials science has matured enough to matter. The remaining challenge isn't whether the field is real. It's whether teams are willing to do the implementation work that turns possibility into operating advantage.
If your team is trying to connect fragmented experimental data, add explainable ML to formulation or property prediction, and turn materials R&D into a tighter design-build-test-learn loop, Polymerize is worth evaluating. It focuses on polymers, chemicals, and advanced materials, with an AI-native system designed to unify R&D data and support model-guided experimentation in enterprise settings.

