A familiar scene plays out in materials R&D every day. A formulation chemist has a target property in mind, a shortlist of raw materials, and a stack of past experiments spread across spreadsheets, ELN entries, instrument exports, and slide decks. The team runs another round of trials, adjusts one ingredient, changes one process condition, and hopes the next sample moves in the right direction.
Sometimes it does. Often it doesn't.
That cycle is expensive because materials systems are rarely simple. Properties emerge from interactions between composition, processing, structure, and testing conditions. Human intuition still matters, but intuition alone struggles when the design space gets wide and the data history gets messy. That's where machine learning in materials science starts to matter in a practical way. Not as a replacement for scientists, and not as a black box that magically invents better materials, but as a way to learn from accumulated experimental history and make the next experiment more deliberate.
The companies getting value from this aren't the ones chasing flashy algorithms first. They're the ones that treat data quality, validation, and interpretability as part of the scientific workflow. They connect what happened in the lab to what the model can credibly predict, then use that model to narrow options, expose trade-offs, and prioritize experiments worth running.
Table of Contents
- Why most lab data is harder to use than teams expect
- What an AI-ready materials dataset actually contains
- A practical sequence for getting started
- Translate ML terms into lab terms
- Forward prediction is not the same as inverse design
- What different learning modes are good for
- Start from the decision you need to make
- Where Gaussian processes fit
- Why tree ensembles are common in industrial work
- A model that isn't validated isn't ready for lab decisions
- Interpretability is part of model governance
- What trust looks like in practice
- Formulation screening when historical data is scattered
- Process scale-up when lab success doesn't transfer
- Closed-loop experimentation for faster learning
- Pick one problem with economic weight
- Build the operating model, not just the model
- Measure scientific and business outcomes together
Introduction From Trial and Error to Targeted Discovery
Traditional materials development often runs on a pattern that feels scientific but behaves like managed guesswork. A team starts with prior knowledge, adjusts a few formulation variables, runs tests, reviews results, and repeats. That works when the system is simple and the number of interactions is small. It breaks down when a polymer blend, additive package, process window, and target performance profile all influence each other at once.
The frustration usually isn't a lack of effort. It's a lack of visibility. Teams can't easily see which variables matter most, which combinations are worth testing next, or where the historical data already contains useful signal.
A major shift began in the 2010s with the rise of data-driven materials informatics. Researchers started using historical experimental and computational datasets to predict phase diagrams, crystal structures, and material properties at scale. A NIST review of machine learning in materials science notes that machine learning methods were already being used for accelerated prediction of phase diagrams and crystal structures, development of interatomic potentials and energy functionals, and high-throughput analysis of experiments. That matters because it marks the point where ML moved from a promising idea to a practical part of discovery workflows.
Where the old approach stalls
In enterprise labs, the bottleneck usually appears before anyone says “AI.” A scientist wants to answer a business question like these:
- Property targeting: Which formulation is most likely to hit viscosity, adhesion, and cure requirements together?
- Failure reduction: Which combinations are likely to be unstable or out of spec before we spend lab time on them?
- Scale-up risk: Which process parameters are tightly linked to performance drift when moving from bench to pilot?
Without a predictive layer, teams often run broad experimental matrices and prune them manually. That can still produce good science. It just does so slowly.
Practical rule: Machine learning is most useful when the number of plausible experiments is much larger than the number you can realistically run.
What machine learning actually adds
Machine learning in materials science works best as a computational partner. It learns patterns from prior formulations, process settings, characterization results, and outcomes, then estimates what's likely to happen for new candidates. Used well, it doesn't eliminate experimentation. It makes experimentation more selective.
That changes the nature of R&D work. Instead of asking, “What should we try next based on instinct alone?” teams can ask better questions:
- Which candidates have the highest predicted chance of success?
- Where is the model uncertain, meaning new data would be especially valuable?
- Which raw materials or process conditions seem to be driving the prediction?
Those are operational questions, not academic ones. They connect directly to cycle time, experimental efficiency, and technical confidence. The rest of the challenge is building an enterprise workflow that can support them.
Unifying Your Foundation The Data Strategy for Materials AI
Most machine learning projects in materials fail for ordinary reasons. The data is fragmented, naming conventions aren't consistent, test conditions are missing, and no one agrees on which result is the trusted one. Teams often discover that their biggest obstacle isn't model choice. It's assembling a usable record of what the lab has already learned.
Why most lab data is harder to use than teams expect
A single formulation program might spread information across recipe spreadsheets, ELN pages, instrument software, PDFs from external labs, LIMS records, and email attachments. Each source can be useful on its own. Together, they're hard to align.
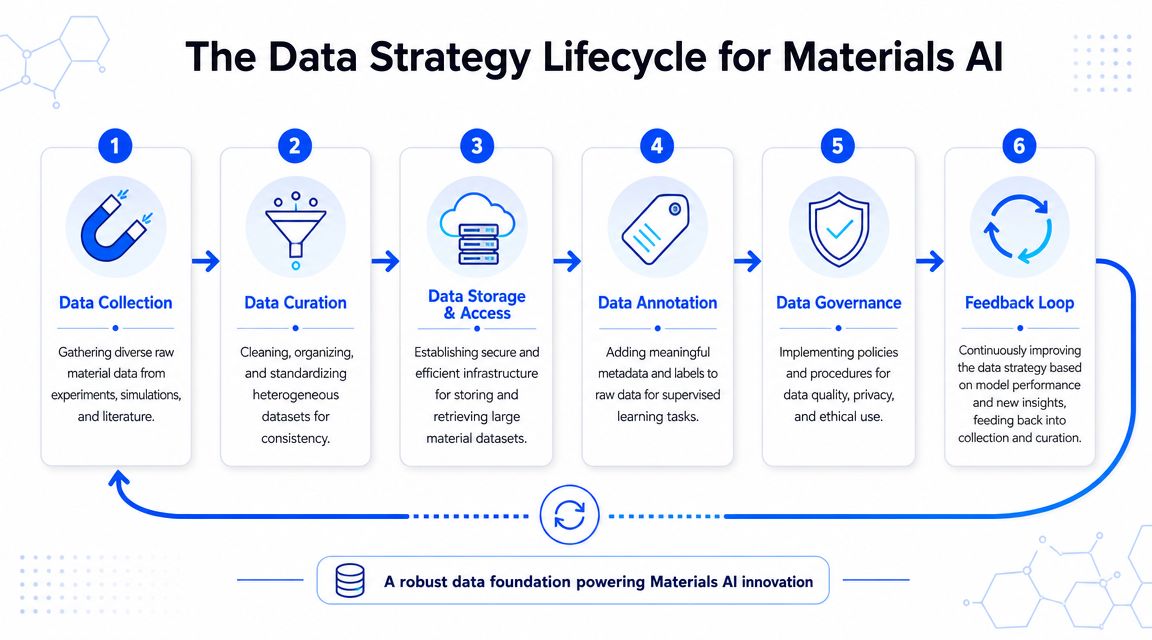
The first discipline is to stop thinking in terms of files and start thinking in terms of entities and relationships. For materials work, that usually means linking:
- Material identity: Raw materials, grades, suppliers, lots, and substitutions
- Formulation structure: Component names, concentrations, order of addition, and intended function
- Process history: Mixing conditions, temperatures, dwell times, cure schedules, line settings
- Characterization context: Test method, specimen prep, environmental conditions, operator notes
- Outcome data: Properties, pass/fail decisions, observations, defects, and stability outcomes
If one of those layers is missing, the model can still run. But the scientific value drops quickly. A tensile result without specimen prep context, for example, may be mathematically valid and scientifically misleading.
What an AI-ready materials dataset actually contains
AI-ready doesn't mean perfect. It means structured enough that the same experiment can be interpreted the same way every time. In practice, that requires a few choices that teams often postpone for too long.
A useful materials dataset usually includes a controlled vocabulary for ingredients and processes, standardized units, explicit treatment of missing values, and metadata that preserves experimental context. Image data and spectra can also be valuable, but they need labels tied back to formulation and process records.
The fastest way to stall a materials AI program is to train on outcomes without preserving how the sample was made and tested.
Document ingestion matters here too. Many historical records live inside reports, PDFs, and scanned technical packages. If your team is extracting formulation tables or test results from those sources, tools that automate document processing with pdf-parser can help convert unstructured records into something the data team can curate instead of retyping by hand.
A practical sequence for getting started
Don't begin by trying to centralize everything the organization has ever produced. Start with one product family, one property family, or one decision bottleneck.
A workable sequence looks like this:
- Scope one decision path. Pick a question such as formulation ranking, failure classification, or process troubleshooting.
- Identify source systems. List every place the relevant data lives, including side files and instrument exports.
- Normalize the basics. Resolve units, names, IDs, and duplicate records before deeper modeling work.
- Capture metadata. Add method, condition, and provenance fields so the data stays scientifically interpretable.
- Create access rules. R&D, process, and analytics teams need shared access patterns without exposing sensitive IP broadly.
- Set a refresh loop. New experiments should flow back into the dataset routinely, not through occasional cleanup projects.
A platform approach often helps because it gives the organization one operational backbone instead of many disconnected repositories. Polymerize is one example of a system built to unify experimental records from spreadsheets, ELNs, and lab silos into a centralized data layer for materials R&D. The important point isn't the vendor name. It's that the enterprise needs a durable data backbone before predictive work can scale.
Core Concepts The Language of Materials AI
A typical failure pattern in enterprise materials AI starts with language, not algorithms. The lab team is discussing formulations, process windows, and failure modes. The data team is discussing features, labels, and model classes. If those terms are not mapped cleanly, projects drift into translation work instead of solving an R&D decision.
Translate ML terms into lab terms
In a materials program, features are the variables used to describe a sample, formulation, or process run. They can include monomer ratios, additive loading, particle size, solvent class, extrusion temperature, cure time, humidity during testing, or descriptors derived from molecular structure.
A label is the outcome the team wants to predict or classify. That may be tensile strength, conductivity, viscosity, adhesion score, glass transition behavior, or a pass/fail result.
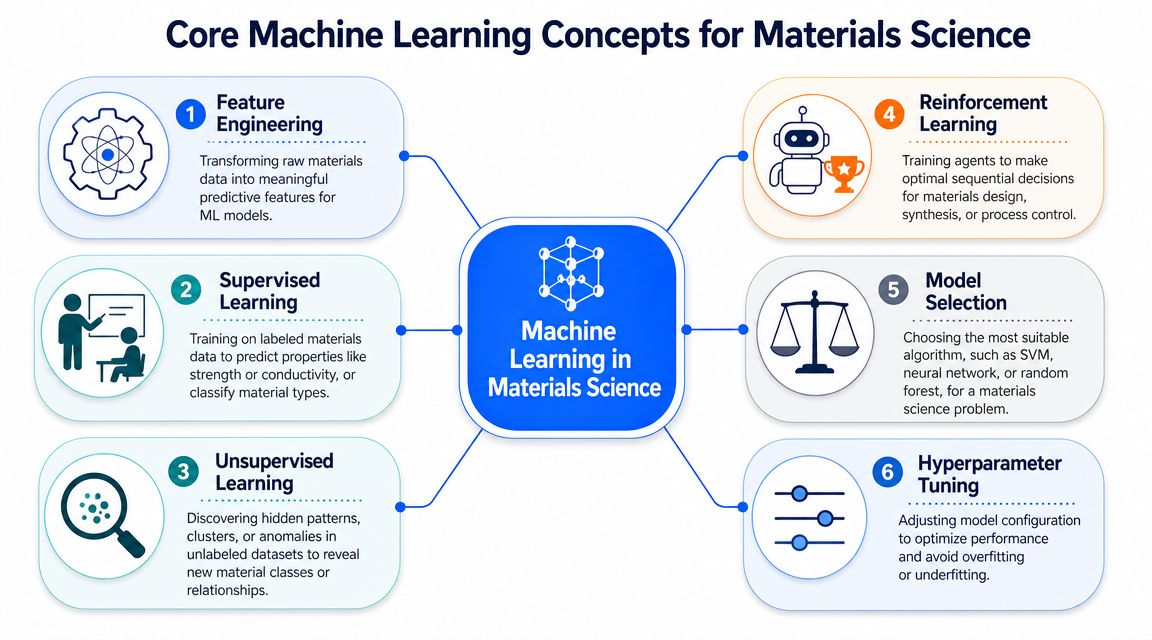
Feature engineering is where raw experimental history becomes model-ready input. This step often determines whether the model reflects the science or just reflects whatever happened to be easy to export from instruments and spreadsheets. A raw process log usually needs to be converted into variables with physical meaning, such as peak temperature, time above a threshold, cooling rate, shear history, batch age, or interaction terms between composition and process conditions.
In practice, domain judgment proves its worth.
Two teams can start from the same ELN records and build very different datasets. One will dump columns into a model and get unstable results. The other will encode the experiment in a way that preserves mechanism, constraints, and context. The second team usually reaches useful predictions faster, even with less data.
Forward prediction is not the same as inverse design
These terms are often blurred together in vendor pitches and internal strategy decks, but they support different decisions.
Forward prediction asks: given this formulation and process recipe, what properties should we expect? That is the more common industrial starting point because it supports screening, ranking, and experiment prioritization.
Inverse design asks: what formulation or process settings are likely to meet a target property profile? That problem is harder because many candidates can satisfy the same target on paper, while still failing cost, manufacturability, stability, regulatory, or scale-up constraints.
| Task | Typical lab question | Practical challenge |
|---|---|---|
| Forward prediction | What happens if we make this candidate? | Needs reliable historical input-output data |
| Inverse design | What should we make to hit these targets? | Needs constraints, search logic, and feasibility checks |
For most enterprise R&D groups, forward prediction is the better first milestone. If the model can rank candidates well enough to reduce low-value experiments, the team already has a business case. That same pattern shows up in adjacent industrial use cases where companies cut downtime with machine learning by predicting outcomes early enough to change operations.
What different learning modes are good for
Machine learning methods are usually grouped by the kind of feedback available during training.
- Supervised learning: Use this when historical records include known outcomes and the goal is to predict a property or class. Most formulation-property and process-property work sits here.
- Unsupervised learning: Use this when outcomes are missing or inconsistent, but the team still wants to find clusters, hidden structure, or unusual runs. This is useful for segmenting material families, comparing process regimes, or flagging anomalous experiments for review.
- Reinforcement learning: Use this in sequential optimization problems where each decision affects the next experiment. It is less common in day-to-day enterprise programs, but it matters in closed-loop experimentation and some autonomous lab settings.
The point of this vocabulary is operational clarity. Scientists need to know what question the model is answering, what data it requires, and where it can fail.
A model is not reasoning about chemistry the way a senior formulator does. It is estimating statistical relationships between how a material system was described and how it behaved under specific conditions. Once teams align on that, discussions about data quality, experiment design, validation, and deployment become far more productive.
Choosing Your Toolkit Common ML Models and Their Jobs
No model is universally right for materials R&D. The better question is which model fits the decision, the dataset, and the way scientists need to use the output.
Start from the decision you need to make
If the target is a continuous property such as modulus or viscosity, you're in a regression setting. If the target is pass/fail, defect/no defect, or stable/unstable, you're in classification. That part is straightforward.
The actual selection criteria are usually these:
- Data volume: Is the dataset small and expensive, or broad and continuously growing?
- Uncertainty needs: Do scientists need to know where the model is unsure?
- Interpretability: Will the team trust a prediction without seeing the drivers behind it?
- Nonlinearity: Are there many interactions between composition and process variables?
- Operational use: Is the model ranking candidates, flagging failures, or guiding the next experiment?
That's why teams rarely settle on one model family forever. The toolkit changes with the maturity of the program.
Where Gaussian processes fit
For small, expensive datasets, Gaussian Process Regression is often a strong choice because it provides both a prediction and an uncertainty estimate. That uncertainty is useful when experiments are costly and the design space is only partially sampled. Instead of only asking which candidate looks best, the team can also ask where the model lacks confidence and where a new experiment would teach the system the most.
That's especially relevant in early-stage formulation work, alloy screening, and polymer discovery, where every lab run has a real cost and sparse data can make overconfident extrapolation dangerous.
Why tree ensembles are common in industrial work
When datasets become larger and the interactions become more nonlinear, tree-based methods are often more practical. The industrial perspective on AI and machine learning in materials science from Polymerize describes Random Forest, XGBoost, LightGBM, and CatBoost as offering a strong balance of predictive performance and interpretability, with SHAP used to extract feature attributions. The same source notes a common two-track strategy: GPR for small-data, uncertainty-driven planning, and boosted trees for scalable prediction plus explainability when datasets are larger.
That two-track pattern matches what many practitioners see in the field. Early programs want uncertainty. Mature programs want throughput, reliability, and explanations that scientists can audit.
| Model | Primary Use Case | Handles Small Data? | Provides Uncertainty? | Interpretability |
|---|---|---|---|---|
| Linear models | Baseline regression, simple directional effects | Sometimes | Not inherently | High |
| Random Forest | Property prediction, classification, nonlinear effects | Often reasonably | Limited by default | Moderate |
| XGBoost | High-performance structured data prediction | Usually better with more data | Limited by default | Moderate with SHAP |
| Gaussian Process Regression | Small-data prediction and experiment planning | Yes | Yes | Moderate |
| Neural networks | Complex pattern learning with richer datasets | Usually not ideal at the start | Not inherently | Lower without extra methods |
A model that predicts well but can't influence experimental decisions is still only half useful.
For adjacent manufacturing use cases, it's worth looking at how reliability teams cut downtime with machine learning. The domain is different, but the lesson carries over. Model choice should follow the operational decision, not the other way around.
Validation and Interpretability Trusting the Black Box
A formulation team has three candidate experiments left this week, one shared pilot line slot, and a model that ranks the options in an order the senior chemist does not expect. That is the moment when validation and interpretability stop being academic. The question is no longer whether the model is mathematically interesting. The question is whether the organization should spend time, material, and production capacity based on its output.
Trust in materials ML is built the same way trust in any lab method is built. Teams check whether it works under realistic conditions, whether its failure modes are understood, and whether the result can be tied back to something scientifically coherent.
A model that isn't validated isn't ready for lab decisions
A close fit to historical data says very little by itself. In enterprise R&D, the true test is whether the model can rank or predict outcomes for experiments, lots, or process conditions it has not seen before.
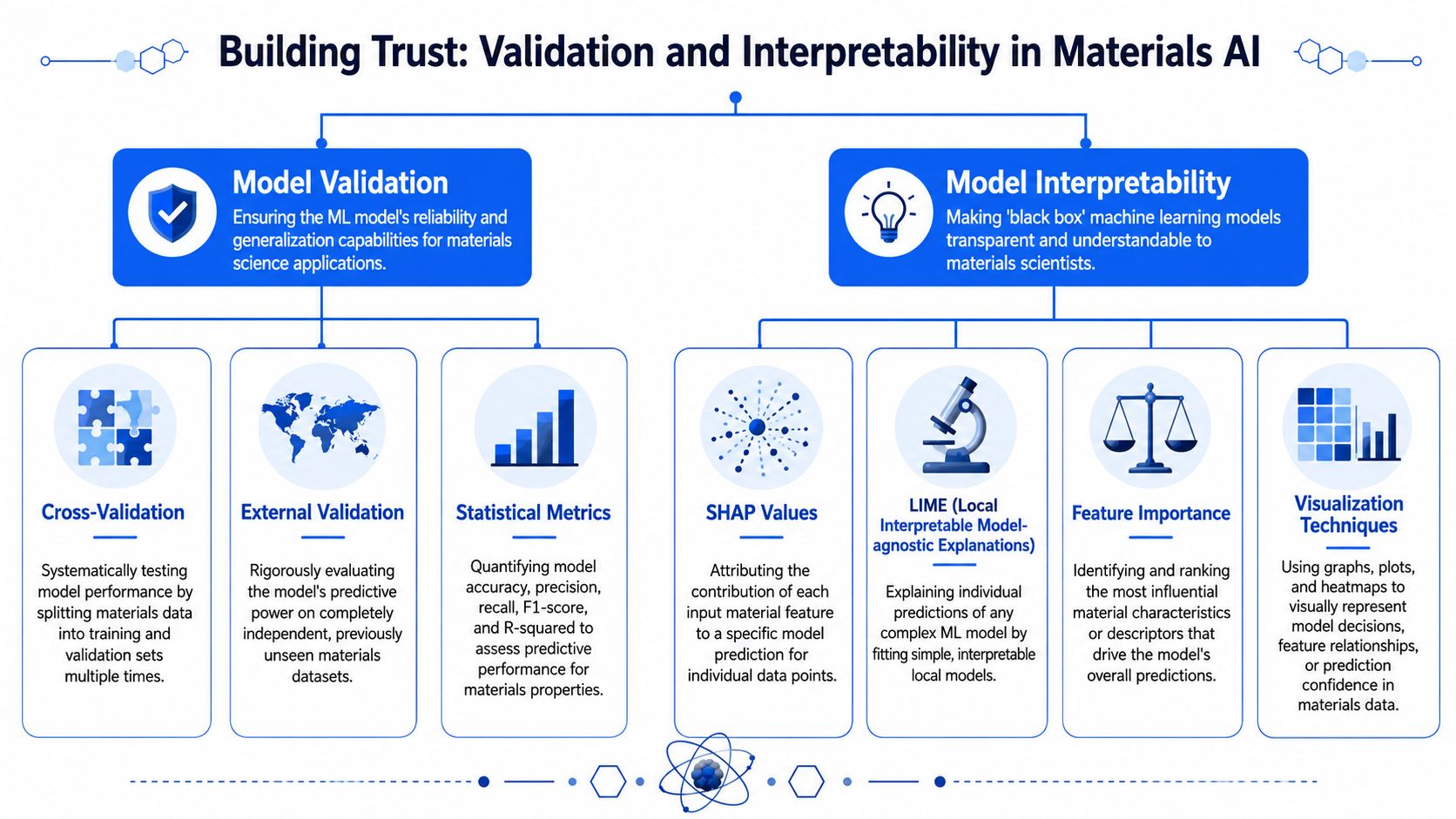
That sounds straightforward, but materials programs rarely operate on clean, independent samples. Data often arrives in clusters from the same campaign, instrument, operator, or raw material lot. If those near-duplicates are split carelessly between training and test sets, reported performance looks better than the model will deliver in the lab.
The metrics also need to match the decision. For classification problems, teams often track measures such as precision, recall, F1-score, and ROC-AUC. For property prediction, common choices include R2, mean absolute error, and root mean squared error. Cross-validation helps when datasets are small, but it does not replace a protected test set that is held back until model development is largely complete.
A practical validation workflow usually includes:
- A protected test set: Keep it untouched until model selection and tuning are finished
- Cross-validation during development: Use it to compare candidates and estimate variability in performance
- Domain-aware splitting: Separate by batch, campaign, composition family, time period, or scale when those factors could leak information
- Baseline comparison: Check whether the model improves on simple heuristics, historical averages, or a linear baseline
- Failure review: Examine the misses by chemistry, process window, and measurement method, not only by summary metric
I have seen teams approve a model with acceptable average error, then lose confidence after a few visible misses in a chemically important region. That is usually a validation design problem, not just a modeling problem. If the holdout set does not reflect the decisions the lab will make, the metric is answering the wrong question.
Interpretability is part of model governance
Interpretability matters because materials scientists need to judge whether a prediction is useful, suspicious, or outside the model's experience.
For tree-based models, feature attribution methods such as SHAP can show which inputs pushed a prediction up or down. For linear models, coefficients can expose directional effects quickly. For Gaussian processes, uncertainty can be as informative as the prediction itself. None of these methods proves causality. They do something more practical. They show whether the model's reasoning is at least consistent with known physics, process knowledge, and measurement constraints.
That matters in an enterprise workflow.
A model may appear accurate because it learned a proxy variable tied to test order, a reformatted sample ID, or a measurement artifact from one instrument. Interpretation methods help surface those shortcuts before they get embedded in screening or scale-up decisions. In regulated or quality-sensitive environments, that review is also part of governance. Teams need to document why a model was trusted, where it should not be used, and what signals would trigger retraining or retirement.
Field note: Adoption improves when explanations help a scientist decide the next experiment, not when they only satisfy curiosity about the algorithm.
What trust looks like in practice
In a working R&D program, trust is observable. Scientists know when to use the model, when to ignore it, and when to ask for more data.
| Trust element | What it looks like in practice |
|---|---|
| Scientific plausibility | Key drivers are understandable to subject matter experts and do not conflict with basic mechanism knowledge |
| Statistical discipline | Evaluation uses held-out data, repeatable splits, and metrics tied to the business decision |
| Boundary awareness | The team can identify extrapolation, sparse regions, and conditions that fall outside training coverage |
| Operational fit | Predictions arrive in time to influence experiment planning, formulation screening, or process troubleshooting |
| Auditability | Assumptions, data lineage, model version, and known limitations are documented |
When these pieces are in place, the model stops being a black box in the unhelpful sense. It becomes a decision tool with known strengths, known limits, and a clear role in the path from fragmented lab data to experiments worth running.
Real World Case Studies ML in Action
The most convincing use of machine learning in materials science isn't a benchmark chart. It's a workflow that changes what the lab does next.
Formulation screening when historical data is scattered
A specialty materials team often begins with a familiar problem: years of experiments exist, but no one can query them cleanly. Formulation recipes sit in spreadsheets. Test outcomes live in PDFs. Process notes remain buried in notebooks and local files. Scientists know there's signal in the record, but extracting it for one project takes more time than running a fresh experiment.
The practical first step isn't advanced modeling. It's unifying enough historical data to answer one narrow question, such as which candidates are most likely to fall within an acceptable performance window. Once the data is cleaned and linked, even a modest supervised model can help rank candidates and identify regions of formulation space that look promising or risky.
The value here is operational. Teams stop treating every new request as a fresh search problem.
Process scale-up when lab success doesn't transfer
A second common case appears during transfer from bench to pilot or pilot to manufacturing. The formulation may be sound, but the process window isn't. Mixing order, thermal history, shear conditions, or residence time can alter the final structure enough that the same nominal recipe behaves differently at larger scale.
In this setting, the model's job is less about discovering a new material and more about separating critical parameters from noisy ones. Engineers can combine formulation variables with process history and final quality outcomes, then use an interpretable model to see which settings appear tightly linked to drift or failure. The benefit is that scale-up discussions become less anecdotal.
Some of the best ML projects in materials don't invent a new formulation. They tell the team which process variables are quietly undermining a good one.
A short explainer helps make that point concrete:
Closed-loop experimentation for faster learning
The field is also moving beyond retrospective prediction. A recent MRS tutorial positioned Gaussian Processes and Active Learning as tools for autonomous experimentation and autonomous phase mapping, showing that ML is increasingly being used to choose the next experiment rather than only analyze previous ones, as noted in the earlier section's cited materials informatics literature.
That shift matters in real programs because it changes the rhythm of experimentation. Instead of designing a full matrix up front, teams can run a smaller batch, retrain, and let the model recommend where the next data point should come from based on predicted value and uncertainty.
Closed-loop systems aren't appropriate for every lab. They require data discipline, instrumentation readiness, and scientists who are willing to work with model-guided iteration. But when those conditions are in place, the workflow becomes much more targeted. The model stops being a report generator and becomes part of experimental planning.
Deploying ML in Your Lab Best Practices for Enterprise R&D
A common enterprise scenario looks like this. The lab has years of formulation data, process logs, and characterization results spread across LIMS exports, spreadsheets, instrument files, and slide decks. Leadership asks for AI. Six months later, the team has a model demo, but no clear decision point, no owner, and no change in experimental throughput.
Deployment starts with a narrower question. Which decision is expensive, repeated often, and still handled with too much trial and error?
Pick one problem with economic weight
The best first use case sits inside an existing R&D workflow. It should have a defined user, a measurable outcome, and enough historical context to support model training and review. In practice, that usually means targeting a decision that already slows the lab down or consumes scarce pilot, synthesis, or testing capacity.
Good starting points include formulation ranking, root-cause analysis for quality drift, pass/fail screening, and process parameter optimization within a stable product family. Poor starting points tend to be broad transformation efforts with no decision owner, no operational boundary, and no agreement on what success looks like.
A practical shortlist for first projects:
- Experimental prioritization: Rank which candidates deserve scarce lab time
- Failure prediction: Flag likely dead ends before materials are synthesized
- Process sensitivity mapping: Identify which manufacturing variables deserve tighter control
- Knowledge reuse: Reuse historical learning across similar chemistries instead of restarting each project
Build the operating model, not just the model
In enterprise materials programs, deployment breaks down less often on model performance than on process design. Scientists, process engineers, informatics teams, and data scientists need a shared way of working. That includes common data definitions, record-quality rules, retraining criteria, and agreement on what counts as a usable model output.
The handoff into lab work needs to be explicit.
Someone has to decide which predictions appear in the scientist's workflow, how uncertainty is shown, when a recommendation is strong enough to influence experimental planning, and how exceptions are documented when domain knowledge overrides the model. If those choices stay informal, the model usually ends up in a separate dashboard that people review after the final decision has already been made.
The adoption hurdle usually isn't algorithmic complexity. It's whether the model fits the way scientists make decisions.
Measure scientific and business outcomes together
A model can score well in validation and still miss the point operationally. Enterprise R&D teams need evidence that the system improves cycle time, cuts avoidable experiments, sharpens scale-up choices, or increases reuse of prior project knowledge across adjacent chemistries.
That trade-off matters. A more interpretable model with slightly lower predictive performance may be the better deployment choice if scientists trust it, challenge it productively, and use it to change experiment selection. In contrast, a higher-performing model with poor usability often stalls at the pilot stage because no one wants to defend its recommendations in a project review.
A practical deployment checklist looks like this:
| Area | What good looks like |
|---|---|
| Problem framing | One defined decision, one owner, one measurable workflow impact |
| Data readiness | Unified records with formulation, process, and outcome context |
| Validation | Held-out testing, cross-validation, and error review before deployment |
| Interpretability | Explanations that scientists can inspect and challenge |
| Workflow integration | Predictions delivered where the lab already works |
| Learning loop | New experiments flow back into the training set routinely |
Teams that get durable value from machine learning in materials science treat it as part of R&D infrastructure. The goal is better experimental judgment at scale, with faster learning across portfolios and fewer wasted cycles between data collection, modeling, and the next lab decision.
If you're evaluating how to operationalize this inside polymers, chemicals, or advanced materials R&D, Polymerize is one option to review. It combines a centralized data backbone for experimental records with explainable modeling workflows designed for property prediction, formulation optimization, and next-experiment planning in enterprise materials teams.

